本文作者:雲也
今天,可能是阿爾茲海默病藥物研發歷史上,裡程碑式的一天。
美國食品和藥物管理局(FDA)在確定驗證性試驗證實臨床益處後,將衛材用於治療成年阿爾茲海默病患者的 Lecanemab「轉換為傳統批準(traditional approval)」[1]。
這意味著該藥正式通過瞭所有必要的臨床試驗,證明其對大多數目標人群的安全性和有效性,獲得完全批準,可以被廣泛用於醫療實踐。

圖源:美國 FDA 官網
此前,美國 FDA 曾於 1 月 6 日,在安全性和有效性尚未完全證實的情況下,基於其降低患者大腦淀粉樣蛋白沉積的治療效果,「加速批準」Lecanemab 上市。
「加速批準」這一途徑適用於「醫療需求未得到滿足的嚴重疾病急需的藥物」,允許在臨床療效證據不完善前,根據藥物對替代終點的影響,合理預測臨床益處對患者的臨床益處之後批準藥物先期上市。
有美國科技媒體在報道中表示,這是 20 年來 FDA 首次完全批準一款阿爾茲海默病藥物。(「轉為傳統批準」即為獲得瞭「完全批準」)
半月打 1 針,延緩疾病進展 27%
阿爾茲海默病( Alzheimer''s disease,AD)具體病因尚未完全闡明,但其病理特征明確,即 AD 患者的大腦出現 β 淀粉樣蛋白斑塊的水平升高、Tau 蛋白和神經纖維纏結形成,最終導致神經元及其突觸喪失。
此前研究表明,β 淀粉樣蛋白的可溶性寡聚體相比單體有更強的細胞毒性。Lecanemab 能與這些寡聚體特異性結合,促進它們清除。
Lecanemab 本次獲批的關鍵依據,是其 Ⅲ 期臨床研究 Study 301 (CLARITY AD)。該研究是一項多中心、隨機、雙盲、安慰劑平行對照研究,此前在 NEJM 已正式發表 [2]。

圖源:NEJM
研究共納入 1795 名 AD 患者,均處於輕度認知障礙或輕度癡呆階段,同時通過腦脊液或 PET 檢測確認其大腦存在 β 淀粉樣蛋白斑塊。患者以 1:1 比例隨機接受安慰劑或 Lecanemab 治療,劑量為 10mg/kg,每兩周給藥一次。
研究結果顯示,Lecanemab 組治療 18 個月後,主要療效終點評分(臨床癡呆評定量表,Clinical Dementia Rating Scale - Sum of Boxes,CDR-SB)相對基線平均下降 1.21 分,安慰劑組相對基線平均下降 1.66 分(評分降低越少表示病情進展越慢)。Lecanemab 與安慰劑兩組相差 0.45 分,具有統計學和臨床意義,相當於延緩疾病進展 27%。
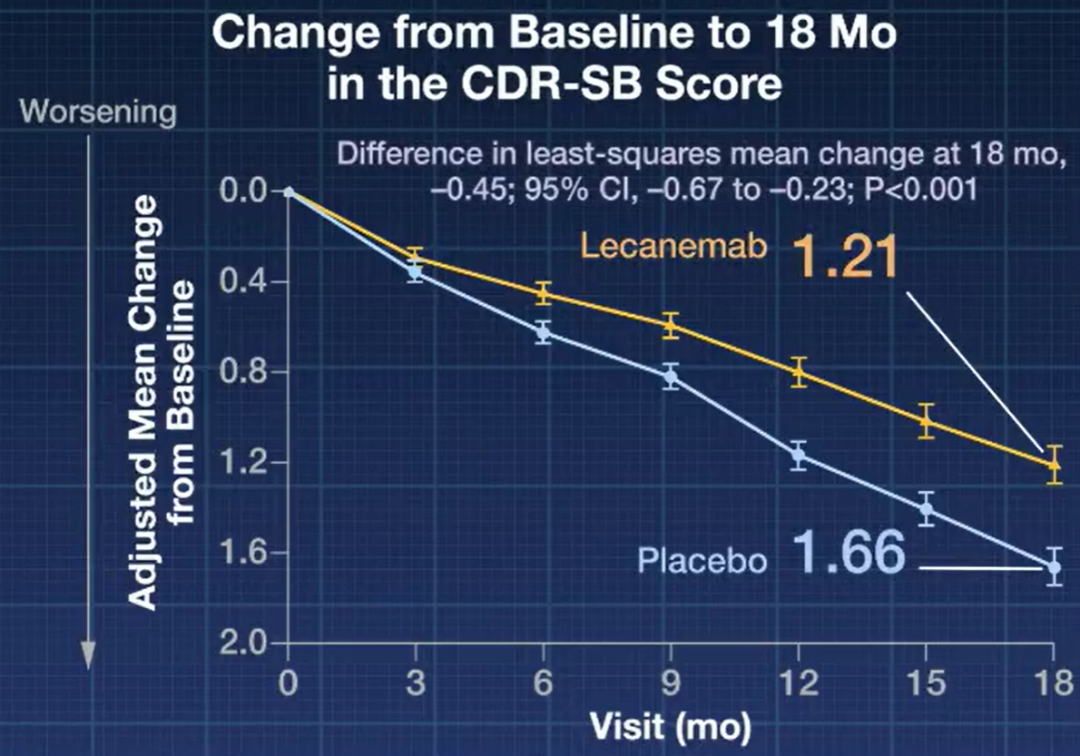
研究主要終點 圖源:NEJM 視頻截圖
同時,在其子研究中,Lecanemab 相比安慰劑,能更大程度地減少大腦中淀粉樣蛋白負荷,差值為 −59.1 centiloids(PET 診斷 AD 的測量單位)。
在安全性方面,美國 FDA 在其新聞中指出,該藥最常見的副作用是頭痛、輸液相關反應和淀粉樣蛋白相關成像異常(ARIA)[1]。
其中,ARIA 最常表現為影像學檢查中出現的大腦局部暫時性腫脹,可伴有大腦內或表面的微小出血點,通常會隨著時間的推移而消退。
該藥處方信息中包含安全性黑框警告,以提醒患者和護理人員註意與 ARIA 相關的潛在風險:少數情況下,ARIA 可能會出現頭痛、意識模糊、頭暈、視力變化和惡心等癥狀,罕見情況下可出現危及生命的腦水腫,伴癲癇發作和其他嚴重的神經系統癥狀;同時須警惕腦出血發生。
ApoE ε4 等位基因純合子患者的 ARIA 風險較高。處方信息要求,在開始用藥治療之前,應進行 ApoE ε4 檢測,並告知患者發生 ARIA 的風險。
20 年來 FDA 首次完全批準
FDA 在新聞中指出,Lecanemab 是第一個從加速批準轉換為傳統批準的阿爾茲海默病 β 淀粉樣蛋白定向抗體。
而上一個加速批準,至今依然深陷質疑。
2021 年 6 月 7 日,美國 FDA 也曾加速批準另一款藥 Aducanumab 用於治療 AD,成為「20 年來首個獲批的 AD 治療藥物」。但在被贊譽為「裡程碑」的同時,質疑也隨之洶湧而來。
兩個至關重要的 Ⅲ 期臨床試驗結果分別於 2015 年 8 月和 9 月相繼啟動,設計完全相同也相互獨立,但兩項試驗均未完成,僅分析瞭部分數據,結果不一致,但又通過亞組分析,才得出瞭高劑量組結果一致的結論 [3]。
試驗結果不一致,產生瞭「臨床獲益不確切」的第一個質疑。同時,FDA 的外部專傢顧問委員會壓倒性反對批準該藥上市,使用從未使用過的「替代終點」,「中樞神經系統 Aβ 負擔減少」,來加速審批該藥上市,這一點備受質疑。
FDA 外周和中樞神經系統咨詢委員會的成員在 NEJM 撰文稱,迄今為止多項基於 β-淀粉樣蛋白假說開發的藥物臨床試驗,「並沒有提供實質性證據能表明 β-淀粉樣蛋白降低意味著臨床獲益」[4]。言下之意,AD 治療中,β 淀粉樣蛋白斑塊減少並不等於臨床有效。
替代終點的有效性受到質疑,意味著這次批準的根基受到動搖。
挫折果然接踵而來。半年後,該藥在歐洲收到瞭一封拒信—— CHMP 投票反對這款備受爭議的藥物獲得監管授權,並在後續的二次審查中主動撤回瞭申請。

圖源:歐洲藥品管理局官網
2022 年 4 月 7 日,美國醫療保險和醫療補助服務中心(Centers for Medicare & Medicaid Services,CMS)更新瞭一項國傢醫保政策,以評估藥物的臨床收益是否值得醫保買單評審結果公示:Aducanumab 的醫保覆蓋將被限制在醫學中心或醫院門診開展的臨床試驗 [5]。
這一決定,不僅意味著 Aducanumab 的醫保報銷嚴重受限,也給其他同類藥物築起一道高高門檻——這項限制同樣適用於其他正在開發的 AD 藥物,包括同公司的 Lecanemab。
即便通過加速審批途徑上市,不意味著藥物研發階段結束,仍需要進行研究以確認預期的臨床益處,目的是為瞭驗證替代終點與臨床獲益的實際聯系,從而證實真正的臨床獲益。如果驗證性試驗表明藥物確實提供瞭臨床益處,則 FDA 會授予完全批準;反之,FDA 將根據監管法規和有條件批準上市要求,將其完全撤下。
Aducanumab 深陷泥淖,而 Lecanemab 則闖瞭過來,拿下瞭 20 年來 AD 藥物的第一個「完全批準」。
6 月 9 日,FDA 周圍和中樞神經系統藥物咨詢委員會(PCNS)全票通過瞭轉為「完全批準」的決議。FDA 藥物評估和研究中心精神科學辦公室代理主任 Teresa Buracchio 表示:「這是首次驗證針對 AD 潛在病理過程的藥物,展現出臨床益處」、「可以確認,對 AD 患者來說這是一種安全有效的治療方法」[1]。
AD 新藥研發領域艱難異常,一些人稱之為「死亡之谷」。
美國制藥行業協會 2018 年 12 月份報告數據顯示,1998~2017 年共 146 個 AD 療法研發失敗。每一個成功上市的 AD 療法背後,都躺著 37 個失敗療法的屍體——成功率 2.7%,低於新藥研發領域的平均成功率 [6]。
Lecanemab 最終完全獲批,即便評審小組成員 Klaus Romero 補充表示獲批之後「仍需要更多研究來繼續驗證該藥的臨床意義」,死亡之谷,終究已有瞭一聲新的回音。
致謝:本文經 復旦大學附屬華山醫院神經內科主任醫師、博士生導師 鬱金泰教授,藥物研發工作者 Klaith 專業審核
策劃:雲也 | 監制:carollero




發表評論 取消回复